
学术资源
STRipy:提高STR异常扩增致病分型的图形应用程序

短串联重复
STR: 短串联重复序列(short tandem repeats,STR)也称微卫星 DNA(microsatellite DNA), 通常是基因组中由1~6个碱基单元组成的一段DNA重复序列。人类基因组DNA中平均每6~10kb就有一个STR位点,不同人体基因组卫星DNA重复单位的数目是可变的,因此,形成了极其复杂的等位基因片段长度多态性。
现有的研究已经充分证明,STR在基因组上的变化与人类遗传疾病之间有着密切的关系。迄今为止,超过 40 种基因的 STR 扩增已被证明会导致遗传性疾病,其中大多数表现出原发性神经或神经肌肉异常。
然而,由于读长限制,短读 NGS 平台无法对大的和/或复杂的重复扩增进行基因分型。本文介绍了一种新的分析软件-STRipy,该开源软件可针对 HTS(high-throughput sequencing) 数据中所有已知的致病性 STR进行分析。
STRipy
STRipy的一大特点是标准和扩展两种模式
当使用“标准”分析时,
仅有STR区域和侧翼2kb区域内的配对reads提交给ExpansionHunter,用于基因分型。
当使用“扩展”分析,
除与STR基因座相邻的区域中提取reads,STRipy还将这些重复reads的比对位置用于识别其它完全重复reads组成致病性重复序列(非靶区L1)。
非靶区L2是那些与非靶向区域L1配对的reads。
该“扩展”分析的最终分析准备文件包括从参考STR基因座周围区域的读取以及从L1和L2非靶区发现的reads。将这些非靶区reads提供给ExpansionHunter,用于基因分型。
STRipy提供了一个STR数据库。
STRipy将ExpansionHunter嵌入后端,并使用数据库将定义的基因座数量从29个增加到55个。STRipy现已包含当前已知的所有致病基因座。
此外,该软件还整合了审查工具REViewer(如图一)来可视化与每个单倍型对齐的reads,同时可视化STR基因座中的变体。
STRipy添加了扩展功能,以实现比测序片段长度更长的等位基因的基因分型。输入文件类型包括BAM(二进制比对图)或CRAM(压缩和参考定向比对图)格式的全基因组、全外显子组或靶向测序比对文件,只要靶向STR基因座上有支持的数据覆盖,即可进行基因分型。
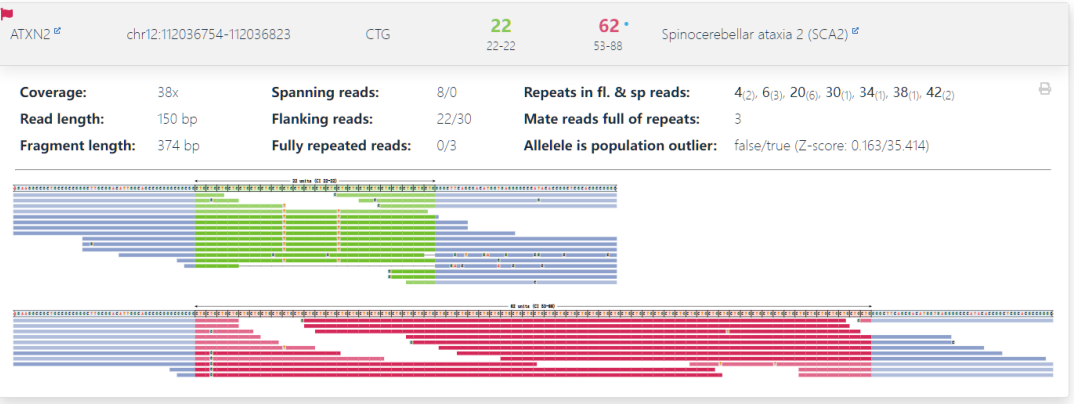
图一、REViewer可视化结果,红色区域:重复数目在致病性的范围;绿色:重复数目正常
STRipy结果展示
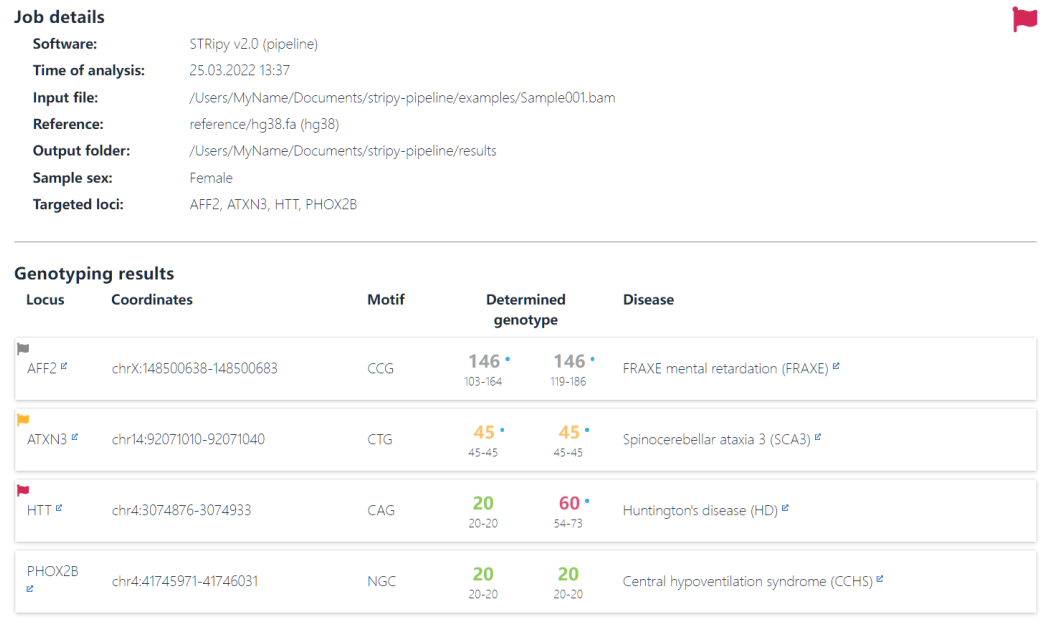
如上图,展示的是一个结果的例子。
红色旗子:重复数目达到致病性范围;
黄色旗子:重复数目在前突变;
绿色:正常范围;
灰色:重复数目范围未知;
重复数目旁边的蓝点:这个重复数目在人群频率中发生的概率是低频,值得关注。
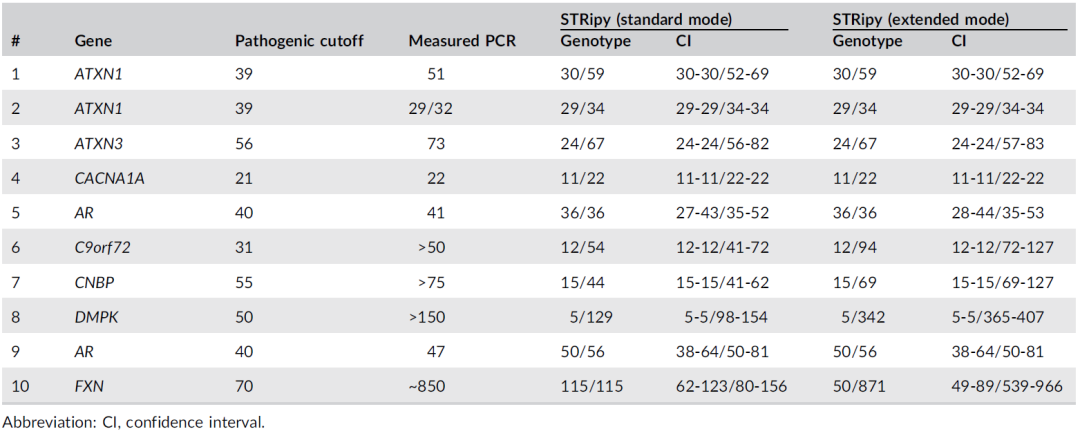
最后,将STRipy应用于一组全基因组测序(WGS)样本验证其有效性,该批次样本先前用于验证STRetch基因分型工具,其中九个样本动态突变类型为致病性,一个样本动态突变类型为前突变。
如下图所示,首先以“标准”模式分析了10个样本(表1),分析结果显示,其中7个样本存在异常的致病性扩增(其中5个样本的估计等位基因长度与PCR测量的长度相同),1个样本为前突变。
对于以“标准”模式分析重复扩增次数小于PCR显示次数的样本,软件会提示并建议使用“扩展”分析。当使用“扩展”分析时,除5号样本外,所有受影响个体的重复扩增数目均达到致病阈值。
参考文献
[1] Halman Andreas,Dolzhenko Egor,Oshlack Alicia,STRipy: A graphical application for enhanced genotyping of pathogenic short tandem repeats in sequencing data.[J] .Hum Mutat, 2022, 43: 859-868.