
新闻中心
融合基因是指两个或多个基因的编码区域首尾相连形成的嵌合基因。染色体上发生的易位、缺失、倒位等结构变异都有可能导致融合基因的出现。随着全外显子组测序(WES)在临床上的广泛应用,利用WES数据识别融合基因变异成了一个新的热点,相关的生信软件也应运而生,本文将对其中几种融合基因检测软件的原理及其优劣进行介绍。

图1 融合基因在染色体上的形成机制
1
FACTERA
FACTERA是2014年发布的一款寻找基因组融合基因断点的软件。该软件使用的输入文件为bam文件,其识别融合的原理分为三个步骤:
✔识别异常的序列簇;
✔检测单核苷酸断点;
✔验证候选融合位点。
具体过程如下:
①序列比对软件(如BWA)在将序列比对至参考基因组时,会对异常序列进行处理,如果某段序列左右两端能同时匹配到基因组的不同位置,软件会对序列进行切割。通过遍历bam文件中的异常序列,识别出一些有着明显断层的异常序列簇(图2 A)。
②找到这些异常序列簇并对异常序列的左右两段序列分别进行处理,左端序列以10碱基为滑窗,并对滑窗内序列做K-mer(K = 4)处理,结果存入HASH表中,右端序列以4碱基为滑窗,得到短序列集合,若HASH表和短序列集合之间是否存在相似性,那么这就是一个潜在的融合位点(图2 C)。
③序列中出现的重复碱基为断点的判定增加了一定的难度,FACTERA默认选择最左侧的位点作为断点。考虑到反向互补序列的存在,融合基因序列在识别过程中,会出现4种方向,但最终呈现时,只有两种方向(GENE1-GENE2,GENE2-GENE1,如图2 D)。在对断点校正之后,FACTERA会使用BLASTN将异常序列簇以及比对至参考基因组失败的序列与生成的融合基因序列进行比对,从而验证该融合位点的准确性。

图2 FACTERA 软件工作原理图
2
GeneFuse
有的研究认为,基于mapping结果识别融合基因有一个缺陷,就是他们过分依赖于mapping软件,如果BWA在mapping过程中未能准确识别所有的异常序列簇,那么极可能导致假阴性。2018年发布的融合基因检测软件GeneFuse,就是一款不依赖碱基识别软件从fastq文件开始分析的融合基因检测软件。
GeneFuse仅关注临床上具有明确致病意义的融合基因,软件内置的靶向融合基因检测区域的数据来源是COSMIC。如果使用者有其他的关注基因,可以修改或自行生成自己的靶向区域配置文件。
GeneFuse检测融合基因的原理可以分为4个步骤:检索、比对、过滤、报告(图3)。
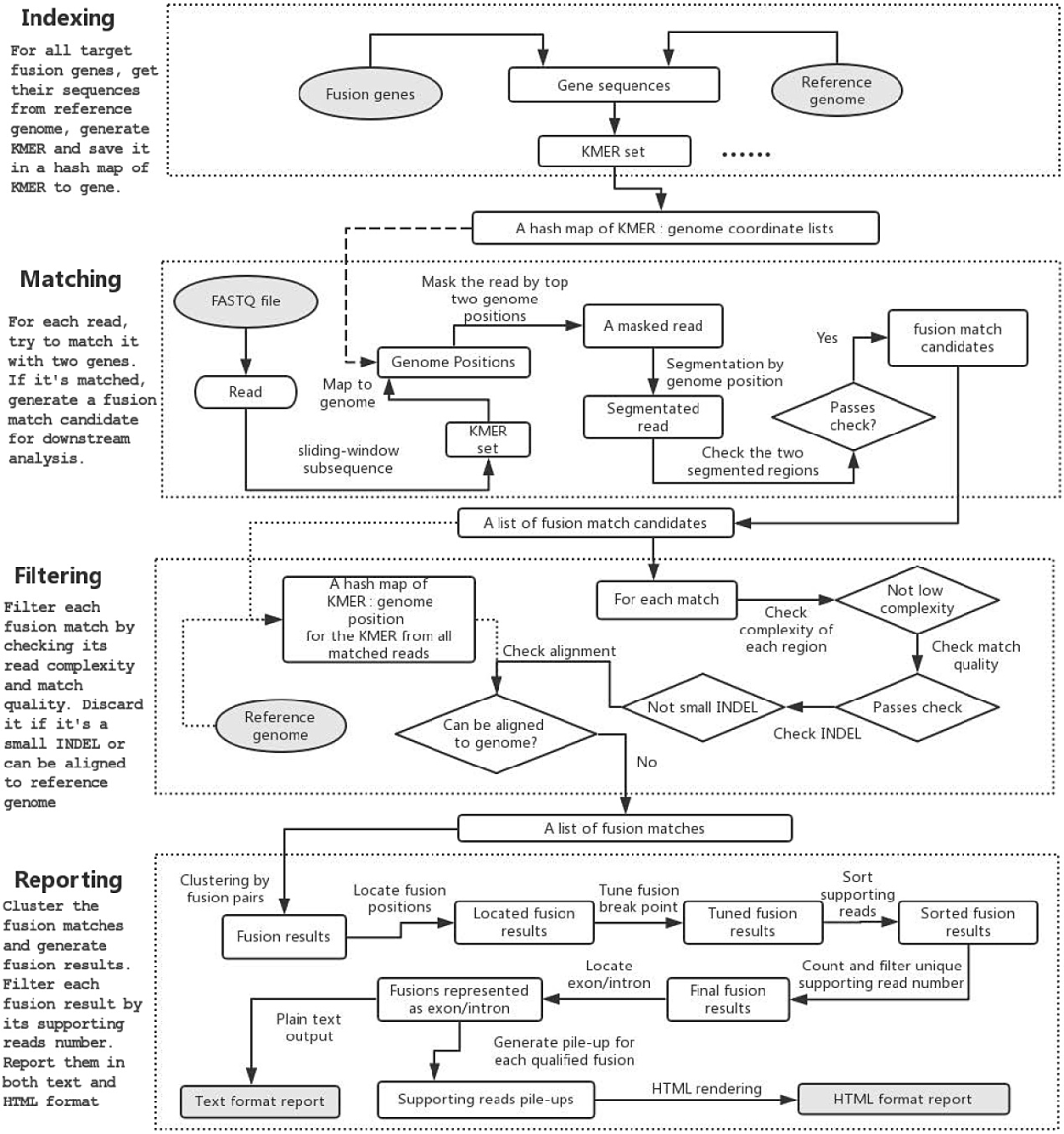
图3 GeneFuse融合基因检测原理图
第一步:在参考基因组上找到靶向基因的参考序列,生成K-mer(K= 16),并以染色体坐标位置作为Key值存入HASH表;
第二步:将输入fastq文件中的序列做k-mer处理,k-mer结果与HASH表进行比对,如果同一条序列能比对到不同基因的HASH表,且左右两段序列均大于20bp,未比对成功的碱基小于10bp,则认为是一个融合基因候选位点;
第三步:进行低复杂度过滤、mapping质量过滤、基因缺失过滤、非独有比对结果过滤;
第四步:将结果根据区段分类,优化断点,合并相同序列,定位外显子并形成txt和html两种格式的报告结果。
3
novoBreak
不管是FACTERA还是GeneFuse,都是利用短序列的比对来检测融合基因,若断点周围和参考基因组差别较大则会受到较多限制。2017年发布的融合基因检测软件novoBreak则将短序列组装为长序列再识别融合基因(图4)。

图4 novoBreak融合基因检测原理图
novoBreak的输入文件为bam文件,将bam文件中的序列切割为k-mer(K = 31)结果,过滤掉所有能够正常比对至参考基因组的序列,找到所有异常序列后,对异常序列前后端进行组装并形成长序列,再将长序列与参考基因组进行比对。如果长序列能一分为二,比对至不同的基因,那么这就是一个融合基因的候选位点。
融合基因的软件多种多样,且识别融合基因的原理各不相同,本文所介绍的三种软件分别采用了不同的原理且各有优劣。
✅FACTERA和novoBreak均使用bam文件作为输入文件,可以使用我们在识别SNV/INDEL、CNV的过程中生成的中间文件再作为输入文件,可以有效节约计算资源和时间成本。
✅FACTERA利用短序列比对,占用内存较小。
✅novoBreak需要组装成长序列,虽然准确性增加,但对内存要求较高。
✅GeneFuse的结果不需要依赖BWA等碱基识别软件的准确性,但是耗时会增加。
如何选择合适的软件来检测融合基因,就需要使用者根据自己的需要自行判断啦。
参考文献
1、Chong Z , Ruan J , Gao M , et al. novoBreak: local assembly for breakpoint detection in cancer genomes[J]. Nature Methods, 2016.
2、Shifu C , Ming L , Tanxiao H , et al. GeneFuse: detection and visualization of target gene fusions from DNA sequencing data[J]. International Journal of Biological Sciences, 2018, 14(8):843-848.
3、Newman A M , Bratman S V , Henning S , et al. FACTERA: a practical method for the discovery of genomic rearrangements at breakpoint resolution[J]. Bioinformatics(23):23.